Clonación del gen de la polifenol oxidasa de aguacate
Este trabajo se basa en una propuesta de una serie de alumnos de cuarto de bioquímica de hace varios años, la cual es claramente una aplicación de los conocimientos de bioquímica adquiridos a lo largo de los años, si bien, el autor de esta entrada de encuentra a penas terminando segundo.
En primer lugar, es necesario contextualizar, exponer la problemática y qué solución se propone, para así entender por qué seguimos los pasos que vamos a mostrar. Esta enzima, la polifenol oxidasa, es una proteína muy extendida en el reino vegetal que se encuentra principalmente en los frutos. La misma y una vez abierto, en este casi el aguacate lleva al pardeamiento típico que observamos, de modo que si logramos inhibir su actividad, lograremos que el aguacate dure más tiempo y por lo tanto, gana interés desde el punto de vista comercial. En este caso, la idea de la que partimos es la inhibición no de la propia enzima, sino de su síntesis por parte del ribosoma, para lo cual aprovecharemos los propios mecanismos de regulación postranscripcional que las células eucariotas poseen, con el empleo de ARN pequeños, en este caso siRNA, ya que son de síntesis exógena. Estos se unen al ARNm de cierta proteína y por complementariedad, total o parcial, bloquean la síntesis de proteínas, por lo que así no habrá expresión de la polifenol oxidasa.
1.Secuencia del gen de la polifenol oxidasa
Es el primer paso, conocer la secuencia que queremos silenciar para así poder diseñar los siRNA. Para ello nos iremos al NCBI, en concreto de la bases de datos de nucleótidos, si bien, nuestra sorpresa es que no se encuentra ninguna polifenol oxidasa de aguacate, de modo que tendremos que hacer una búsqueda de otra forma que hasta ahora no habíamos empleado. Para ello, lo que haremos es buscar el gen de una polifenol oxidasa vegetal, la cual nos sirva como referente a la hora de buscar en el genoma del aguacate, ya que posiblemente la enzima esté conservada. En este caso empleamos el de Malus domestica, o manzana, la cual si se encuentra en la base de datos, la cual tiene como número de acceso D87669.1 y cuya entrada vemos a continuación.
Para seguir, lo que haremos es un BLASTn, donde le pediremos que compare con la secuencia de ADN del aguacate o Persea americana, tal y como se muestra en la siguiente imagen, de modo que podamos localizar en el genoma del mismo al región que posiblemente codifique para la enzima que estamos buscando.
De este mismo encontramos una serie de resultados que se muestran en la imagen siguiente.
De todas ellas, seleccionamos la primera, por presentar mejores valores estadísticos, lo cual aporta fiabilidad. El resultado del alineamiento se puede ver en la captura que se puede ver bajo estas líneas.
Por tanto, ya tenemos la secuencia del gen a silenciar, de todo el genoma del aguacate, es justo este punto el que nos interesa que no se exprese gracias a la técnica que se ha expuesto al comienzo de este trabajo.
2.Amplificación por PCR
El protocolo ya lo estudiamos en la primera práctica del curso, de modo que solo nos centraremos, de forma muy superficial en el diseño de los primers gracias a la herramienta pick primers que nos facilita el NCBI. Seguiremos el mismo proceso que en aquella vez, con los siguientes ajustes:
Con estos el resultado que se proporciona es el que se muestra abajo:
Seleccionamos la pareja nº2 .
3.Diseño del siRNA
Es el siguiente paso, diseñar las secuencias de ARN de silenciamiento que emplearemos en este trabajo. Para ello empleamos una web (OligoWalk) que nos facilita la secuencia ya demás, la idoneidad de cada una, de modo que elegimos el primero de a imagen que a continuación se muestra.
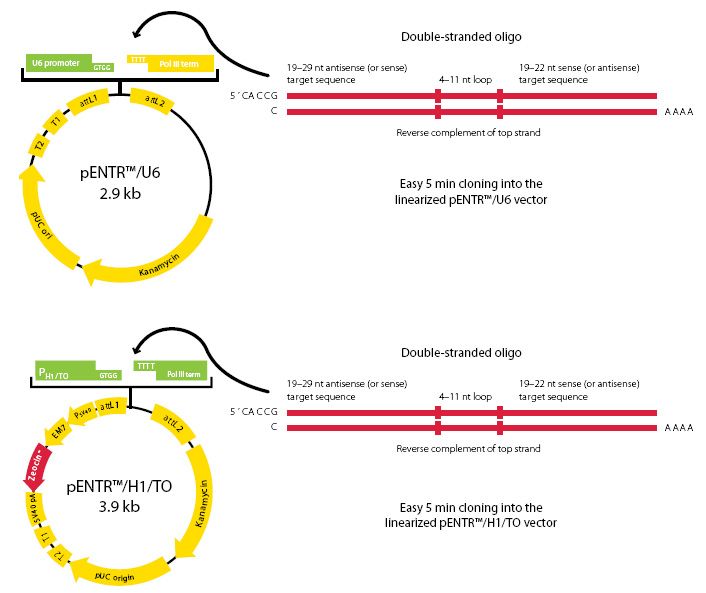
Además de la secuencia hay que introducirlo en la célula, para lo que se usan una serie de vectores, como el que nos facilita la empresa ThermoFisher y cuyo esquema se muestra a continuación. Este vector, intriducido en la célula permite que al expresarse, el ARN que buscamos se sintetice de forma semejante a como lo hace en el mecanismo biológico de regulación del que participa y bloqueará la traducción del ARNm del gen de la polifenol oxidasa.
4.Clonación molecular
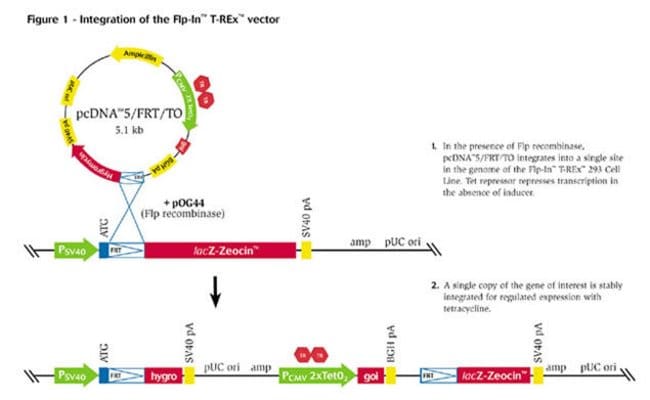
Ahora llega el momento de la clonación del gen de la polifenol oxidasa, de modo que debemos insertar el amplicón de la PCR. Como es un gen de eucariota debe expresarse en células eucariotas y someterse a un splicing, de modo que presentamos el siguiente vector para eucariotas:
Mediante un protocolo de transformación, tenemos que seguir el protocolo que se adjunta (protocolo del vector de expresión). En concreto en este caso, tendremos que emplear un medio con ampicilina y X-gal además de IGTP (un intensificador de la expresión del gen LacZ). Las colonias que sean blancas son las que nos interesan a nosotros, y son de las que replicaremos para obtener un cultivo transformado.
5.Silenciamiento
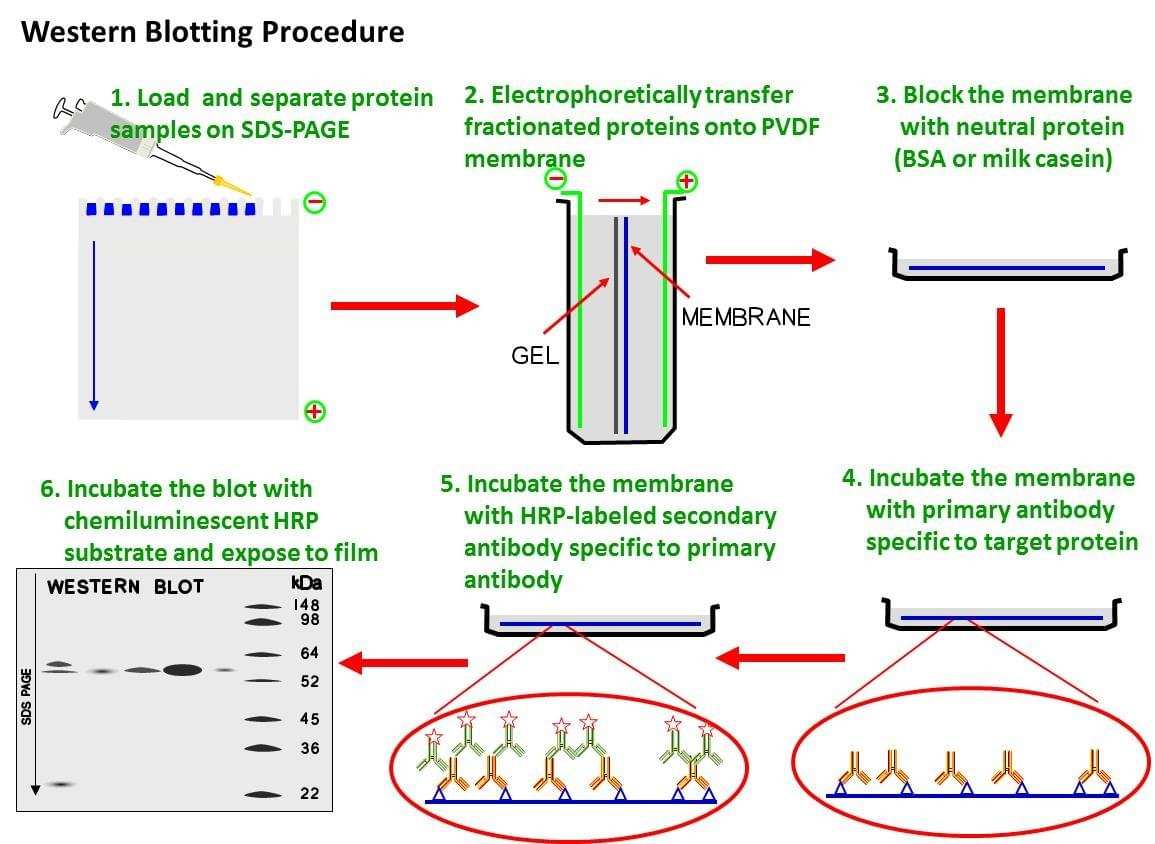
Para ello, transformaremos estas células con el vector de silenciamiento que ya explicamos antes, de modo que, podremso evaluar por blotting la expresión de los genes, gracias a una sonda monolocus, de modo que se hace un proceso parecido al anterior, se toman las colonias transformadas y de estas purificaremos el ARN presente y por un Nothern, hibridaremos con la sonda que diseñaremos y así comprobamos que hay ARNm de la enzima. Posteriormente, por un Western (necesitamos un anticuerpo anti-PPO) podemos evaluar al expresión ( no esperando que haya enzima).
-Nothern blot: se resume en la imagen siguiente.
Primero hay que aislar el ARN de las células (con plásmido del gen y de las silenciadas) para lo que se presenta el siguiente kit de extracción de la empresa Thermofisher del cual se presenta además el siguiente protocolo. Ahora podremos hacer el Nothern, para lo cual hemos encontrado otro kit de la misma empresa y su correspondiente protocolo. En lo que respecta a la sonda, no he logrado encontrar ningún protocolo de síntesis, para l que se me ocurre encargar un oligo o cadena de ADN marcado ya sea por fluorescencia directa o indirecta, del gen que estamos buscando o un fragmento de este, de modo que así hibride con el ARNm y lo detectemos.
Con esto comprobamos la expresión del ARNm en las células sin el vector de silenciamiento, ya que en las silenciadas, habría degradación del ARNm por lo que no se podría detectar, mientras que en las no silenciadas, debe detectarse en la población.
-Western blot: nos servirá como comprobación de los resultados del Nothern Blot anterior.
Se necesita en primer lugar el anticuerpo. En esta web se podría encargar. Después habría que hacer el inmunoensayo, que se resume a continuación:
A la hora de hacer el ensayo, es necesario extraer proteínas, para lo se aporta el siguiente kit y su correspondiente protocolo. Ahora podemos hacer la electroforesis y el inmunoensayo, gracias al siguiente kit siguiendo los pasos que se indican en su protocolo.
El resultado que esperamos es que en las células no silenciadas sí haya detección de la proteína y en las silenciadas que no haya ninguna señal propia de la proteína, ya que eso significa que efectivamente, no se está expresando y que el ensayo ha sido un éxito.
6.Mapa de restricción
Finalmente, se aportan una serie de herramienrtas para diseñar el mapa de restricción del plásmido, lo cual supone saber también si estaba o no insertado el gen de la enzima antes de la transformación de las células. Por ejemplo, el de Biomodel.
CONCLUSIONES
En conjunto, las prácticas del curso y en concreto esta, han supuesto todo un resto, del cual nunca pensé que pudiera salir, ya que no estamos hechos a tener que diseñar por nuestra cuenta una estrategia, sino a seguir paso a paso los protocolos que se nos dan en el laboratorio de prácticas, si bien es normal ya que primero hay que aprender las técnicas y a hacerlas para después poder saber realmente de qué hablamos, ya que el hecho de que casi no haya ido al laboratorio influye, inevitablemente en como enfoco estos trabajos, ya que desconozco trucos o fallos típicos que se aprenden de la experiencia que algún día espero ganar en este ámbito, ya que la bioquímica no es solo teoría, fórmulas y genes, es también manejo del laboratorio, de técnicas y de aplicación de la teoría.
viernes, 1 de mayo de 2020
Corrección de un cromatograma
Si recordamos la práctica anterior, la cual versaba sobre la clonación del gen de la insulina, el último paso fue la secuenciación del gen del propio investigador. El servicio de secuenciación nos devuelve un cromatograma, fruto de la irradiación de la muestra con un láser y que devuelve los picos de emisión de fluorescencia. Sin embargo, hay que evaluar la calidad del mismo y corregirlo.
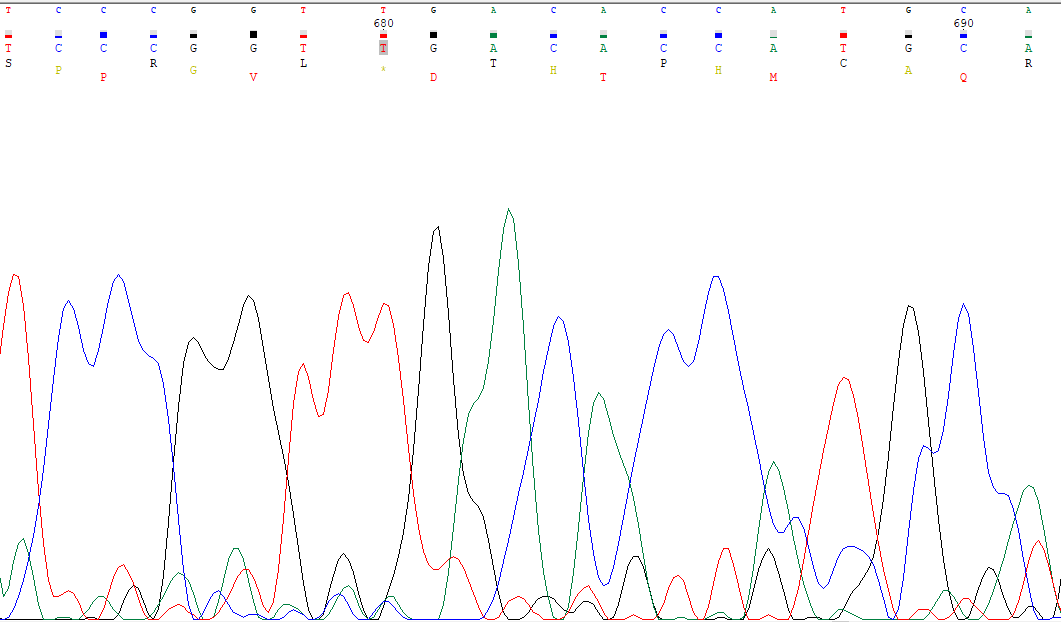
Toda la visualización del cromatograma la hemos hecho empleando el programa Chromas y el archivo empleado es de formato .abi. En un primer lugar, la imagen que vemos sería algo tal que así:
Como se puede observar, el cromatograma nos devuelve los picos asociados a cada base con un colo diferente y en base al pico más pronunciado, se debería haber asignado la base correspondiente. Precisamente, ese debería es la causa por la que nos encontramos aquí: el sistema de secuenciación no es 100% fiable y es necesario que el investigador compruebe la veracidad de lo resultados, teniendo siempre en cuenta que cabe un pequeño margen de error en ciertas decisiones a tomar, como se verá a lo largo del proceso. Lo primero es conocer si la secuencia que hemos recibido ya se ha secuenciado, lo cual nos permitirá comparar la secuencia obtenida y la que se encuentra en el repositorio. Por ello lo primero que hemos hecho es un BLAST-n el cual permite hacer una búsqueda entre todas las secuencias que se encuentran en la base de datos del NCBI que nos permitirá conocer la especie y el que se secuenció. Desde el programa es posible ejecutar dicho BLAST, con el siguiente resultado:
Como vemos, la secuencia "query" es la que nosotros introducimos para comparar y solo las dos primeras nos dan una mayor cobertura de al secuencia, por lo que es muy probable que la secuencia que hemos recibido sea la de dicho gen aún con variaciones. Además de este gráfico, tenemos la tabla con las entradas, que se muestra bajo estas líneas:
Son los datos que corresponden a cada una de las secuencias que se muestran en la imagen anterior, con los valores de e, el porcentaje de cobertura, al putuación etc. Si observamos, la primera secuencia es la que mejor datos nos ofrece con un e-value de 0, lo cual implica que la probalilidad de que el alineamiento se deba al azar es nula y además tiene una buena puntuación (1015) en base a la matriz de sustitución empleada (BLOSUM-62). Al abrirla encontramos la siguiente imagen:
Como podemos ver en la imagen, la cobertura de esta secuencia y las coincidencias es tan buena que podemos asumir que la secuencia que tratamos es el gen de un factor de transcripción, el bHLH75 de Fragaria vesca, en conreto al subespecie vesca, por lo que a partir de ahora buscaremos sobre este organismo. Además, podemos afirmar que la secuencia ante la que nos encontramos ya se ha secuenciado, puesto que encontramos este entrada con una coincidencia elevada.
Obtenido el organismo, el siguiente paso de determinar sobre qué parte del cromatograma vamos a poder trabajar, puesto que de todas las bases que hemos obtenido secuenciaas no todas son de calidad, puesto que como se ha dicho, hay fallos en el proceso de secuenciación. Hay que delimitar dos extremos, tanto el margen izquierdo como el derecho. Si nos fijamos en la imagen primera de este trabajo, no se eligió al azar. Marca el paso de una zona de alto ruido (no es posible determinar bien los picos y otra zona muy limpia, donde claramente se puede definir las bases. Además, en el primer BLAST-n se nos confirma que en torno a la base 161-162 es la secuencia coincide, por lo que seleccionamos como límite izquierdo la base 162 en este caso. En cambio el otro margen no queda tan claro, ya que la región que marca el BLAST-n como límite es un tanto confusa en nuestro cromatograma como se puede ver a continuación.
Ahora hacemos otro BLAST-n, delimitando la secuencia tal y como llevamos hecho hasta ahora, buscando específicamente en Fragaria vesca. El resultado es el siguiente:
Sería complicado afirmar que esas bases son todas correctas en lo que a los picos respecta aún coincidiendo con el cromatograma, por lo que necesitamos un criterio para decidir. Este será la discrepancia que no podamos corregir porque haya duda en la decisión. Buscando entre estas, la que encontramos se localiza en la base 691, donde hay un hueco en la secuencia que tenemos, ya que la secuencia del GenBank hay cuatro guaninas, como se muestra a continuación.
En el cromatograma apreciamos lo siguiente:
Por tanto, no podemos afirmar cuál es correcta ya que no hay solo cuatro posibles guaninas, sino cinco, por lo que es aquí donde en un principio marcaremos el límite derecho, en la base 691, siendo esta la última que se queda justo antes de este problema.
Ahora es momento de corregir las N (bases que se han detectado pero no se les ha asignado una base), encontrando que no hay ninguna dentro del margen de la secuencia que hemos delimitado. Por ello acompañamos de una corrección como ejemplo. En este caso apreciamos que se corresponde a una guanina, pues vemos un pico de color negro que aunque es más bajo que los de su alrededor, se ve claramente. Por otra parte podemos comprobar en el BLAST-n
Podemos contrastar la información con el BLAST-n que hicimos, donde apreciamos que efectivamente, esa base es una guanina, de modo que habría mejorado la calidad del BLAST-n si tuviese en cuenta esta región. Se muestra la imagen del BLAST-n a continuación:
A lo largo de la secuencia encontraremos puntos donde haya dudas, ya que no todos los picos son tan claros como cabría esperar, mostrando el siguiente ejemplo:
Se aprecian 3 claros picos de guanina, pero hay uno que no queda completamente claro (marcado en azul) si es un pico y se corresponde con una base o es simplemente un hombro de la curva. Ante esta duda podemos adoptar dos soluciones: tomar la idea de que, según nuestro criterio hay una cuarta guanina o no, o la opción que vamos a tomar, irnos al BLAST-n y comparar si en esa posición hay o no 4 guaninas. En el mismo encontramos el siguiente resultado:
Vemos que la secuencia devuelta por la base de datos, solo hay tres guaninas y no cuatro como podríamos sospechar, por lo que esta es la base de la decisión a tomar: ese posible pico no es más que el hombro de la curva, por lo que no se corresponde con ninguna base. Es necesario indicar en este punto que la secuencia que usamos como referencia procede de otra secuenciación, por un tercero, por lo que no sabemos los errores que se pudieron cometer a la hora de hacer dicha secuencia, así pues aceptar lo que esta dice siempre entraña asumir cierto riesgo, que en este caso parece aceptable ya que nosotros no tenemos clara la respuesta a la pregunta.
Sin embargo, hay que analizar también la traducción de la secuencia, ya que el resultado final es el de una proteína, por lo que la similitud con esta puede aportar nueva información sobre la calidad del cromatograma. Al igual que en el caso anterior, podemos hacer un BLAST-x, el cual se hace sobre la proteína resultante. El resultado de muestra a continuación:
La secuencia está muy bien pero hay un punto marcado con un asterisco, que simboliza un codón de STOP, de modo que teóricamente si la secuencia fuese correcta veríamos que la proteína resultante no sería tan larga como la silvestre. Es necesario ir a cromatograma y analizar qué ocurre, si podemos mejorar lo que tenemos o cambiar el corte y asegurar más. Lo que si que podemos afirmar que de los tres marcos de lectura posibles el correcto es el segundo, aunque, como esto depende de dónde hemos cortado podría haber discrepancia, por lo que comprobaremos cual es el correcto en el programa. Para ello hacemos que se muestren los tres marcos de lectura posibles como se ve en la imagen siguiente, correspondiente a la zona discrepante.
Como vemos, efectivamente el marco de lectura es el segundo, ya que se da la secuencia correcta. Por otro lado, se comprueba que es un codón de stop el que se codifica en esa zona (TGA). Una posible solución sería intentar evitar dicho codón de stop, para lo que analizamos qué podríamos cambiar (siempre congruente). Este posible cambio sería añadir el pico que, según parece, no se detectó. Si se hace dicho cambio, vemos que hay un desfase y por tanto la proteína resultante se ve alterada:
En consecuencia, esta zona muestra una indecisión clara, por lo que, es conveniente dejarla fuera de la región que consideramos es correcta. Así pues el cromatograma qudaría con el límite derecho en la base 674. Para comprobar si esto ha hecho que la calidad del BLAST aumente, repetimos ambos, tanto n como x, cuyos resultados mostramos a continuación.
En lo que al BLAST-n respecta, vemos como la calidad ha mejorado, pasando el porcentaje de identidad de 97 a 98% y los huecos (gaps) al 0%, de modo que la corrección es desde el punto de vista del BLAST-n muy buena.
Por otra parte, en el BLAST-x ocurre parecido, pasando del 94% de identidad al 98%, un claro salto de calidad. También vemos que las diferencias que hemos encontrado entre BLAST-n y secuencia del cromatograma que deducimos, no podíamos cambiar por lo definido del pico, son SNP )Single Nucleotide Polimorfism) si no superan el 1% de distribución entre la población o mutaciones, pero en todo caso, silenciosas ya que el cambio en el genoma no se ve reflejado en el fenotipo gracias a la degeneración del código genético, que amortigua las variaciones inducidas en el genoma.
Práctica primera: Clonación del gen de la insulina humana
En esta página se pretende presentar y explicar el protocolo a seguir para la secuenciación Sanger de un fragmento de DNA que contiene el gen de la insulina humana, empleando como material de partida el DNA genómico contenido en células sanguíneas, en concreto las células denominadas glóbulos blancos, ya que los glóbulos rojos o eritrocitos carecen de material genético en detrimento de la presencia de hemoglobina (hemoproteína que permite el transporte de oxígeno) y las plaquetas o trombocitos carecen de núcleo debido a que proceden de una célula precursora mayor denominada megacariocito.
En primer lugar, se debe extraer el material genético de las células contenidas en la sangre del individuo, para lo cual encontramos de una forma completamente accesible una serie de kits preparados con tal fin, los cuales contienen todos los reactivos necesarios para obtener DNA en las condiciones apropiadas para realizar una amplificación del genoma por PCR, lo cual se presentará con posterioridad en esta misma página.
En Internet es posible encontrar varios kits disponibles en las webs de muchas casas comerciales, encontrando además los protocolos de los mismos. Todos estos permiten que a partir de pequeñas cantidades de sangre y sin necesidad nada más que de una punción en el dedo por ejemplo para obtener un volumen de microlitros, obtener el DNA genómico compatible con la amplificación por PCR, lo cual es clave ya que en pasos posteriores se realiza una PCR. Entre otros, se han encontrado:
De entre todos los mencionados, se ha seleccionado el comercializado por la empresa Sigma Aldrich, llamado GenElute™ Blood Genomic DNA Kit Protocol (NA2010, NA2020), debido a que este incluye entre las distintas soluciones que presentaremos a continuación, la solución tris-EDTA, la cual se refiere en otros protocolos de otras casas comerciales, pero no se proporciona o al menos no se refiere de forma explícita en el protocolo. Esta sustancia es clave para trabajar con sangre, evita que la sangre coagule debido a la pérdida de contacto con el endotelio del vaso sanguíneo, el cual actúa como factor anticoagulante en condiciones fisiológicas, en colaboración con otras sustancias de naturaleza variada como la heparina que mantiene la sangre en estado líquido.
A continuación se procede a explicar o a destacar los pasos a seguir, de modo que para más información se puede ver el protocolo que se facilita en el enlace asociado a nombre del mismo (resaltado en negrita).
En primer lugar existen unos pasos previos, si bien antes se ha de comprobar que en las distintas disoluciones proporcionadas no existen precipitados. Posteriormente se prepara un baño de agua o un termobloque a 55ºC y se preparan algunas soluciones que se van a emplear en pasos venideros.
Extracción, con una lanceta se pincha en la yema de un dedo y se deja caer sangre, (con unas gotas será suficiente) para almacenar en el tubo con la solución de EDTA . Esta debe haber alcanzado antes de su uso la temperatura ambiente, ya que permite que se mezcle mejor.
Posteriormente se prepara la sangre, de modo que se añade un volumen de 20 µL de la proteínasa K en un tubo de microcentrífuga, en el mismo que se añaden 200 µL de sangre de la disolución de EDTA.
Al añadir 200 µL de la solución de lisis C y aplicarle fuerza mediante un vórtex durante 15 s se procede a la lisis de las células contenidas en la muestra de sangre, de modo que entre todos los componentes celulares, que conforman el debriz celular que nos resulta inútil en este protocolo, queda el ADN genómico presente en el interior de los leucocitos y que es el objetivo de esta extracción. Esta se calienta en el termoboque o en agua a 55ºC durante 10 min, lo cual ayuda a desintegrar las envueltas celulares.
Se añaden 500 µL de la solución de preparación de columna a cada uno de los GenElute Miniprep Binding Column y se centrifuga a 12000 x g por minuto. La solución empleada en este proceso aumenta la unión del ADN a la membrana en y por tanto se obtiene un mejor rendimiento.
Se añaden 200 µL de la solución de etanol (95-100%) al lisado obtenido del tratamiento de la muestra con la solución de lisado.
Tras la dilución de la solución de pre-lavado y la solución de lavado concentradas con etanol se añaden 500 µL de cualquiera de las dos soluciones (como la muestra de sangre es fresca se emplea de forma opcional la solución de pre-lavado) y se centrifuga durante 1 min a ≥6500 x g . Se elimina el fluido que queda fuera del centrifugado y se coloca la columna en un nuevo tubo.
Se añaden 500 µL de la solución de lavado a la columna y se centrifuga durante 3 min a una velocidad máxima de 12,000–16,000 x g con el fin de secar la columna. Es importante que esta columna quede libre de etanol antes de diluir el ADN, por ello en caso de que quede algún resto de etanol, se centrifuga la columna durante un minuto más a la velocidad máxima. Se deposita la columna en un nuevo tubo de 2 mL.
Para eluir, es decir, extraer el ADN de la membrana a la que se encuentra unido, se pipetean 200 µL de la solución de elución (10 mM Tris-HCl, 0.5 mM EDTA, pH 9.0) , directamente en el centro de la columna. Acto seguido se centrifuga por un minuto a ≥6500 x g, con el fin de conseguir una correcta elución, que puede ganar eficiencia si se incuba durante 5 min a temperatura ambiente la columna tras la adición de la solución de elución y aplicar los mismos pasos que se han referido ya.
Tal y como se refiere en el propio protocolo, se puede comprobar la calidad del ADN extraído, mediante un análisis espectrofotométrico y una electroforesis en gel de agarosa. Para ello se diluye el ADN en una solución buffer TE (10 mM Tris-HCl, 1 mM EDTA, pH 8.0–8.5) para medir la absorbancia a 260 nm (debe estar ebtre 0,1 y 1,0 unidades de absorbancia), 280 nm y 320 nm en una microcubeta de cuarzo. El fin de la medida a 320 nm es corregir el fondo de absorbancia de 1,0 a 260 nm correspondiente a 50 µg/mL de ADN de doble cadena. con estas unidades se mide la calidad por la aplicación de una ecuación: [A(260)-A(320)]/[A(280)-A(320)], valor que debe estar en el rango de 1,6 a 1,9.
Mediante la electroforesis en gel de agarosa se comprueba el tamaño del ADN obtenido. Un gel al 0,8% de agarosa en el buffer de TBE permite una correcta electroforesis . Para visualizar el material genético es posible emplear bromuro de etidio para poder medirlo después por comparación con un marcador de ADN como ADN lambda Hind. En el resultado final debe observarse una migración única y observarse una banda de alto peso molecular .
Como bien sabemos, en todo ese ADN extraído se encuentra, concretamente, el gen que buscamos, correspondiente a la insulina. El genoma humano presenta 3400 millones de pares de bases, una secuencia tan extensa de la cual solo queremos aislar un fragmento. En primer lugar es necesario conocer la secuencia concreta del gen de esta hormona. Gracias a la bases de datos GenBank, del NCBI, es posible encontrar la secuencia de esta, obtenida por cierto laboratorio. Haciendo una búsqueda y aplicando una serie de filtros, encontramos una secuencia con código AH002844.2, denominada "Homo sapiens insulin (INS) gene, complete cds". Como se puede leer en la anotación del gen, esta secuencia es de 4969 pares de bases, siendo una ínfima parte de la totalidad del genoma humano.
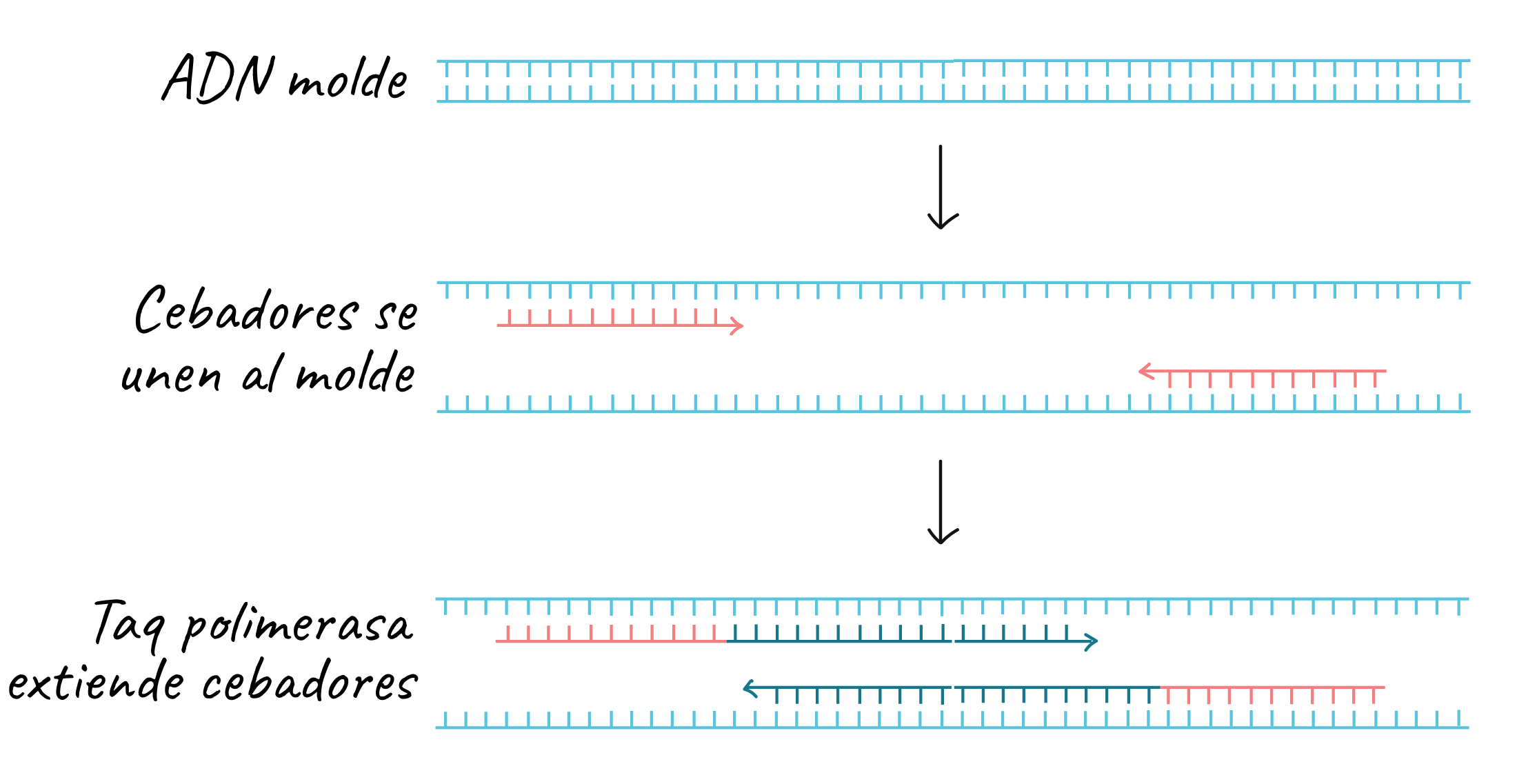
Obtenida la secuencia del gen, podemos, desde la misma entrada, obtener la secuencia de los primers o cebadores de PCR. La función de estos es la de servir como los moldes de la ADN polimerasa, ya que esta alarga la cadena naciente en sentido 5'-3' y lo hace siempre a partir de un extremo 3' (necesita el grupo hidroxilo del carbono 3' del anillo de azúcar. En el proceso biológico de la replicación, la enzima primasa sintetiza sin nucleótido de partida un fragmento de ARN complementario a la cadena molde de ADN. Este presenta unas pocas bases, las suficientes como para establecer una complementariedad específica con cierta región del ADN. Gracias a este cebador de ARN sí hay un donador de OH-3' por lo que a partir de este es posible sintetizar la hebra nueva de ADN, tal y como se muestra en la imagen siguiente, en la cual se pone como ejemplo la propia PCR.
En la propia web del NCBI se da la opción de crear unos primers para la reacción de la polimerasa de la PCR, de tal manera que estos sean específicos de la región del ADN correspondiente al gen de la insulina, el objetivo de esta actividad. Con el fin de simplificar la tarea, haremos tan solo 3 parejas de primers, ya que el proceso de vuelve muy largo y nos centramos en ejemplificar cómo se hace. Dentro de la página del NCBI para el diseño de primers, encontramos el siguiente formulario.
Para este caso, nos basta con esta pantalla para poder obtener los primers, si bien existen muchas más opciones para ajustar la búsqueda a los parámetros del investigador. Iniciamos la búsqueda de los cebadores para nuestra PCR:
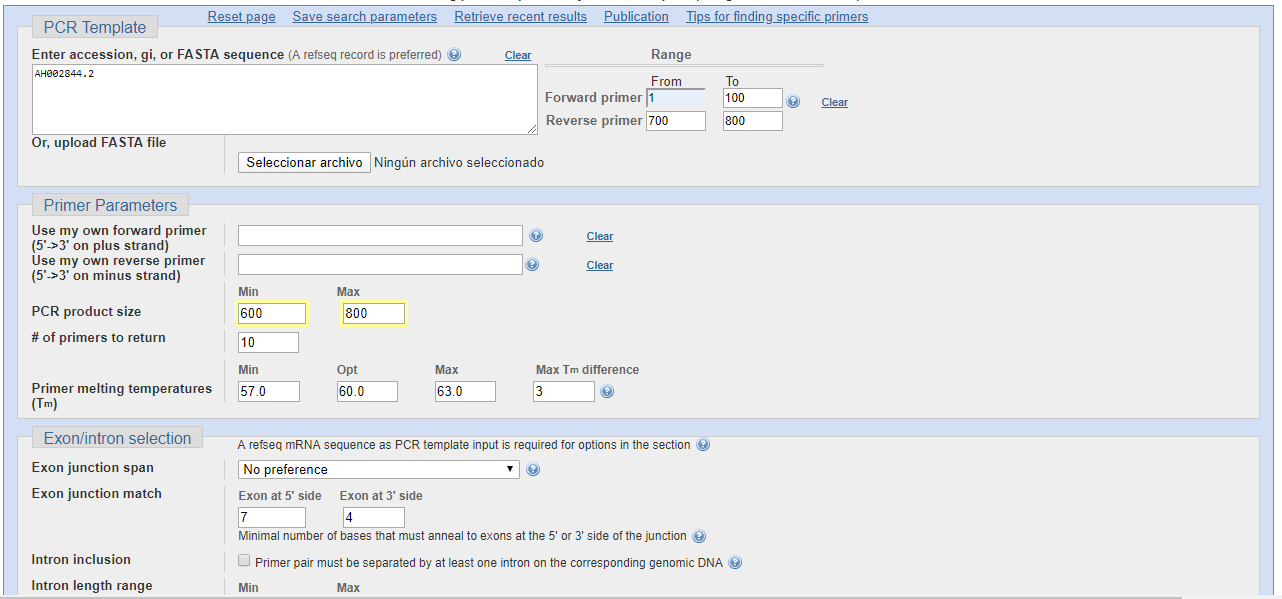
En primer lugar se introduce la secuencia de ADN sobre la que se va a trabajar, de modo que se puede introducir la secuencia copiada, un archivo FASTA o, como hacemos en este caso, la introducción del número de referencia que le otorga el NCBI al gen de la insulina.
A continuación, elegimos las posiciones sobre las que debe encontrarse el inicio del primer. Para ello partimos de que, para hacer compatibles los productos de la PCR con la secuenciación Sanger, optaremos por que los productos de la misma sean de longitudes secuenciables, de modo que no deberán sobrepasar las 800 bases, si bien este número ya es difícil de alcanzar. por ello, para la primera búsqueda, el primer de la cadena denominada como "foward" que va en sentido 3'-5', va desde la base 1 hasta la 100, es decir que como máximo el cebador tomará hasta la base 100. Lo mismo ocurre para la cadena complementaria, para la cual se pone en "reverse" que sea desde la base 700 hasta la 800 como máximo, de modo que el tamaño máximo del producto fuese de unas 800 bases aproximadamente.
Seleccionamos el tamaño de los productos de PCR, que será de 600 a 800 bases, de modo que, como se ha referido, sea compatible con la secuenciación Sanger.
Aplicando estos filtros, tal y como se ve en la imagen anterior, obtenemos una serie de resultados de búsqueda, de entre los cuales hemos seleccionado la pareja nº5, que es la que se muestra a continuación.
Junto con el resultado, se adjuntan una serie parámetros sobre el cebador obtenido, entre los cuales encontramos:
Las secuencias, tanto del foward como reverse y la longitud de las mismas. Esta longitud no debe ser la misma, pero en este caso coinciden. A mayor longitud, se pone de manifiesto una mayor especificidad por la secuencia del ADN a la que debe unirse y por tanto, aumenta la fiabilidad del ensayo.
Longitud del producto de PCR, una 774 bases, lo cual lo hace compatible con el método de secuenciación a emplear.
Mediante Template strand, se hace referencia a cual de las dos cadenas se une, siendo la cadena positiva y la cadena negativa.
Indica, por una parte la base de inicio para cada uno y por la otra la base última incluida en el cebador.
Temperatura de fusión (melting temperature Tm). Este parámetro es crucial para los primers, ya que deben de ser competentes con el ADN de cadena simple de los productos de PCR que se obtienen en la reacción de amplificación que llevamos a cabo. Visto de una forma resumida, esta es la temperatura a la que a mitad del ADN se encuentra unido a los cebadores. El probleama viene cuando, a partir de la tercera reacción ya hay producto de la PCR, el cual interfiere con los cebadores al unirse con la cadena de ADN. Si la Tm del primer es muy baja comparada con la Tm del producto, a las temperaturas que el producto se libera del ADN el primer, debido a la agitación molecular no se uniría al ADN y por tanto la reacción no tendría lugar, de modo que es un parámetro a tener en cuenta aunque en este caso no dé problemas. Se recomienda, eso sí, que Tm(producto)-Tm(primer)<15-20ºC.
Contenido en Guanina, este hace referencia a la proporción de Guaninas que hay en el primer, de modo que estas favorecen la interacción estable ADN-cebador, ya que se unen a la Citosina mediante tres puentes de hidrógeno, frente a los dos de la interacción Adenina-Timina, más débil.
Puntuaciones de auto-complementariedad, esto es la tendencia a formas estructuras como bucles internos.
Continuamos con la siguiente pareja, tal y como se muestra en la imagen siguiente.
Al hacer la búsqueda, encontramos que esta no nos devuelve ningún resultado, sino que nos muestra el siguiente panel informativo.
Mediante una serie de comprobaciones, resulta que en el entorno de las 1400 bases hay una región de baja complejidad, bastante extensa, lo cual hace que la calidad de los posibles primers no sea lo suficiente como para ser compatible con los criterios de búsqueda que hemos dispuesto. Para contrarrestar eso, lo que trataremos es que esas secuencias queden fuera de la región del cebador y que se quede dentro de la región a amplificar, ya que esa baja complementariedad no afecta a la reacción de la Taq-polimerasa. Extendemos el rango cambiando de 1400 a 1300 el inicio del primer reverso.
Ahora sí obtenemos resultados de búsqueda, de entre los cuales seleccionamos la pareja nº2, cuya ficha se muestra a continuación.
Finalmente, para este ejercicio, hacemos una tercera pareja con los parámetros que a continuación mostramos.
De esta búsqueda, seleccionamos la pareja de primer nº10.
Como se ha podido ver, los productos de PCR de cada una de las búsquedas solapan entre sí y esto no es una casualidad: el solapamiento de las secuencias permite que se pueda verificar que la secuenciación ha ido bien, ya que las secuencias se pondrían una sobre otra y así se observa la continuidad de la secuencia.
Ahora podemos pegar la secuencia de cada una de las parejas de primers, de modo que como cada uno de ellos se da en 5'-3', pues no hace falta cambiar el sentido de la misma. A continuación mostramos la confección de una pareja de primers, la primera por ejemplo. Como se puede apreciar en dichas imágenes, se nos facilitan datos sobre los primers que se encargan, en color naranja.
Al terminar, el precio sería, para los cebadores que hicimos antes, de 17,85 €. Como vemos el encargo de los primers es bastante barato, ya que para un primer de 25 bases, el precio sería de 3,75€, cantidad irrisoria dentro del presupuesto de una investigación.
La empresa facilita, en teoría 25nmol de cebadores, pero, en una reacción química (al fin y al cabo la bioquímica es química) las cantidades de reactivos y catalizadores (en este caso, enzimas) son claves a la hora de que la reacción tenga lugar para dar la cantidad de producto deseado. Por ello, es necesario cuantificar la cantidad de cebadores que realmente nos manda la empresa, para lo cual tenemos, por ejemplo, un ensayo de absorbancia, tal y como expone la propia empresa Thermofisher en un enlace específico para la cuantificación de cebadores. Como se manifiesta en ese mismo texto, se diluye la muestra en un factor 1:100 y se mide la absorbancia a 260 nm, ya que los ácidos nucleicos de cadena simple absorben a dicha longitud de onda. Sin embargo, la ley de Lambert-Beer, es necesario conocer el coeficiente de extinción molar (el paso del tubo depende del empleado pero es un dato del propio dispositivo), dato que puede parecer difícil de obtener, pero no es más que la suma de los coeficientes de cada una de las bases que componen el oligonucleótido, secuencia que conocemos. En la tabla siguiente se ven los valores de cada una de las bases y procede de la misma web.
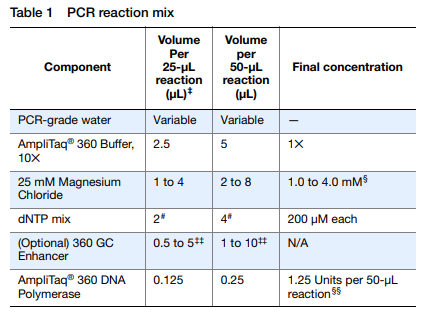
A continuación, es necesario seguir un protocolo de PCR y obtener un kit para la misma, que encontramos en la propia empresa Thermofisher, en el que encontramos un kit llamado AmpliTaq 360 DNA Polymerase. En este protocolo, se nos presentan una serie de tablas donde se indican los componentes, volúmenes y concentraciones de cada uno, tablas que se adjuntan a continuación, de modo que para mayor detalle, se pide visualizar el protocolo y además, a modo de resumen, una línea de trabajo que muestra los pasos a seguir para hacer la PCR.
Si observamos la que se encuentra encima de estas líneas, vemos que se nos pide un volumen de primers para obtener al final, en la solución de PCR la concentración de primers correcta para la reacción, por lo que, es nesario hallarla, para lo que se facilitan a continuación las siguientes cuentas a modo de ejemplo, esto es, el procedimiento modelo, de modo que el fundamento teórico es el mismo independientemente de los valores numéricos. Para este ejemplo tomaremos como volumen a tomar de la disolución madre de primer 2,5µL, ya que 0.5µL es demasiado poco volumen como para tomarlo con precisión. Para ello y tomando como disolución final un volumen total de 25µL, tendremos que realizar las siguientes operaciones para hallar la concentración de la disolción madre de cebadores.
Por tanto, como vemos, los primers deben diluirse hasta una concentración de 10µL para que al tomar 2,5µL se diluyan en el volumen final y se obtenga la concentración de 1µL. Sin embargo, debemos llevar hasta esa concentración los moles de cebadores que nos envía la empresa, supongamos, que son realmente 25nmol que vienen liofilizados, es decir, secos y por tanto no hay agua en ellos. Para ello, habrá que buscar el volumen de buffer que habrá que añadirles para obtener la concentración deseada de 10µL. Para ello, partiremos teóricamente de 25nmol, por lo que como antes, esto es solo una forma de reflejar un fundamento teórico, de modo que jugaremos con valores estándar y en el caso concreto de nuestro ensayo tomaríamos los valores concretos del mismo.
Como vemos, habría que añadir 2,5mL para obtener dicha concentración madre de primers para la PCR. Sin embargo, esta disolución no es exclusiva para una sola reacción de la PCR, de modo que podemos calcular cuantas reacciones de la PCR podemos llevar a cabo teóricamente con esta disolución madre de los cebadores de volumen 2,5mL y molaridad 10µL. Para ello, sabemos que en cada reacción empleamos un volumen de esta de 2,5µL, por lo que solo hay que realizar una sencilla cuenta, tal y como se muestra a continuación.
Por tanto, queda patente como a partir de una sola disolución de 2,5mL podemos realizar 1000 reacciones de la PCR, por lo que, en una situación como la actual, en lo que respecta al coronavirus COVID-19 donde el diagnóstico se hace con PCR, es posible diagnosticar a 1000 pacientes como máximo partiendo de una sola compra de cebadores. Para ejemplificar esto de otra forma, vamos a comprobar sobre la cantidad inicial de moles de cebadores, cuantos gastamos en una sola reacción de amplificación, para lo cual seguimos las siguientes pautas, suponiendo incialmente 25nmol de cebadores y tomando de referencia los valores estándar empleados a lo largo de este apartado.
Observamos que, gastamos el 0,1% del total de moles originales en cada reacción de PCR, de modo que podemos concluir que es bastante rentable desde el punto de vista de los cebadores. Añadimos a continuación una tabla donde se ponen de manifiesto los ciclos de temperatura a los que se debe programar la PCR para que sea efectiva.
A continuación de la reacción de amplificación, es necesario conocer si realmente esta ha sido como esperábamos es decir, si hemos obtenido los fragmentos de longitud sean tal y como hemos programado. Para ello haremos una electroforesis en gel de agarosa, una ténica ampliamente usada en este tipo de pruebas y que nos permitirá verificar que todo ha ido bien.
Para una electroforesis en gel de agarosa lo primero es hacer el gel de agarosa, para lo que incluye un vídeo explicativo donde se muestra paso a paso como se hace y con imágenes del proceso para que quede más claro. Este lo hemos tomado de la Universidad Politécnica de Valencia y lo mostramos a continuación.
Es muy importante el porcentaje de agarosa, ya que eso determinará el tamaño de los poros del gel y por tanto, la selectividad del mismo, pues facilitará el paso de más o menos fragmentos de ADN en base al tamaño del fragmento: mayor tamaño del poro mayor permeabilidad y viceversa. Estimaremos el paso de fragmentos de unas 500 bases (los nuestros son de unas 700 pero para ellos la permeabilidad será menor pero no nula).
Como vemos en esta tabla, el porcentaje recomendado es del 1% de agarosa en la preparación del mismo. Sin embargo, es necesario usar también un marcador, es decir, una muestra patrón que al correr dé un patrón de bandas indicativas de la longitud del segmento de ADN, ya que fragmentos de misma longitud migran la misma distancia sometidos a las mismas condiciones. Así pues, encontramos estos marcadores de tamaño para electroforesis de la casa comercial Ecogen, llamado HyperLadder II, cuto patrón de bandas de electroforesis se adjunta en la imagen siguiente, y sería la referencia para determinar si la reacción de amplificación ha sido eficientes y si tenemos el fragmento que queríamos.
Tras hacer llenar los pocillos y dejar correr las muestras en el gel, podemos seleccionar de cada pocillo la banda que contiene el ADN que nos interesa, los fragmentos de unas 700 bases que queremos enviar a secuenciar. Una opción es purificar dichas bandas, tras extraerlas del gel (cortaremos el cuadrado que contiene dicha banda). Para su purificación se comercialzian kits para la extracción de ADN de los mismos, como el que aquí encontramos, de la casa comercial Merck, presentando este modelo. Adjuntamos el enlace al protocolo del mismo (manual). Como vemos en la imagen de la misma web, es un sencillo proceso.
También, para simplificar la compresión del proceso, encontramos un vídeo que aunque no es nuestro kit, los pasos son semejantes, de modo que ilustra de forma fiable los pasos a seguir.
Obtenido el ADN y teniendo los cebadores para cada uno de los fragmentos que vamos a secuenciar (productos de la PCR), podemos enviarlo a un servicio de secuenciación, como el que se nos ofrece en el SCAI de la Universidad de Córdoba, donde se nos indica cómo se debe enviar el material, tal y como mostramos a continuación.
La tabla mostrada encima de estas lineas ha sido extraída de la propia web del SACAI, del manual de usuario y nos explica cómo debemos enviar las muestras de secuenciación y los cebadores (estos son los mismos que encargamos para la PCR).
Finalmente, adjuntamos una serie de enlaces Web que nos pueden permitir estudiar la secuencia que recibamos, si bien, habrá que corregir el cromatograma que recibiremos del servicio de secuenciación. Dichos enlaces son:
Creación de un mapa de restricción: si queremos expresar la insulina en meidnate recombinación de ADN, es importante saber con qué enzimas podemos restringir el gen y así buscar un vector (tendría que ser de eucariotas debido a la presencia de intrones y exones) donde la misma enzima actúe y así poder unirlos y expresar la insulina en un sistema de expresión.
Reverso complementario, nos puede servir si el gen se expresa en el sentido contrario al que se nos presenta.
Búsqueda de palíndromos: son puntos en los que una posible estructura secundaria es posible por autocomplementariedad.
Intrones y exones: realmente lo que hemos secuenciado es el gen, no hemos determinado de donde a donde van las regiones codificantes que realmente son las que conforman el CDS. Nos facilita un mapa de los mismos.
Fase de lectura abierta (ORF): son las posibles secuencias codificantes que van desde un codón de inicio de la traducción (AUG en el ARN) hasta uno de stop. Posibles lecturas de nuestro fragmento.
En mi opinión, esta actividad ha sido todo un reto: nunca pensé que pudiera hacer ni la mitad de lo que he puesto, ya que hay cosas que son propias de otros cursos según la distribución del grado (por ejemplo las experimentales que acabamos de empezar con MIC). Aún así y a pesar de las reticencias iniciales, es una actividad que permite ganar algo de confianza en un terreno que para mí es temido por la enorme cantidad de técnicas que hay y la falta de base que tenemos. A pesar de ello, estoy muy contento con el resultado del trabajo.