Corrección de un cromatograma
Si recordamos la práctica anterior, la cual versaba sobre la clonación del gen de la insulina, el último paso fue la secuenciación del gen del propio investigador. El servicio de secuenciación nos devuelve un cromatograma, fruto de la irradiación de la muestra con un láser y que devuelve los picos de emisión de fluorescencia. Sin embargo, hay que evaluar la calidad del mismo y corregirlo.
Toda la visualización del cromatograma la hemos hecho empleando el programa Chromas y el archivo empleado es de formato .abi. En un primer lugar, la imagen que vemos sería algo tal que así:

Como se puede observar, el cromatograma nos devuelve los picos asociados a cada base con un colo diferente y en base al pico más pronunciado, se debería haber asignado la base correspondiente. Precisamente, ese debería es la causa por la que nos encontramos aquí: el sistema de secuenciación no es 100% fiable y es necesario que el investigador compruebe la veracidad de lo resultados, teniendo siempre en cuenta que cabe un pequeño margen de error en ciertas decisiones a tomar, como se verá a lo largo del proceso. Lo primero es conocer si la secuencia que hemos recibido ya se ha secuenciado, lo cual nos permitirá comparar la secuencia obtenida y la que se encuentra en el repositorio. Por ello lo primero que hemos hecho es un BLAST-n el cual permite hacer una búsqueda entre todas las secuencias que se encuentran en la base de datos del NCBI que nos permitirá conocer la especie y el que se secuenció. Desde el programa es posible ejecutar dicho BLAST, con el siguiente resultado:

Como vemos, la secuencia "query" es la que nosotros introducimos para comparar y solo las dos primeras nos dan una mayor cobertura de al secuencia, por lo que es muy probable que la secuencia que hemos recibido sea la de dicho gen aún con variaciones. Además de este gráfico, tenemos la tabla con las entradas, que se muestra bajo estas líneas:

Son los datos que corresponden a cada una de las secuencias que se muestran en la imagen anterior, con los valores de e, el porcentaje de cobertura, al putuación etc. Si observamos, la primera secuencia es la que mejor datos nos ofrece con un e-value de 0, lo cual implica que la probalilidad de que el alineamiento se deba al azar es nula y además tiene una buena puntuación (1015) en base a la matriz de sustitución empleada (BLOSUM-62). Al abrirla encontramos la siguiente imagen:

Como podemos ver en la imagen, la cobertura de esta secuencia y las coincidencias es tan buena que podemos asumir que la secuencia que tratamos es el gen de un factor de transcripción, el bHLH75 de Fragaria vesca, en conreto al subespecie vesca, por lo que a partir de ahora buscaremos sobre este organismo. Además, podemos afirmar que la secuencia ante la que nos encontramos ya se ha secuenciado, puesto que encontramos este entrada con una coincidencia elevada.
Obtenido el organismo, el siguiente paso de determinar sobre qué parte del cromatograma vamos a poder trabajar, puesto que de todas las bases que hemos obtenido secuenciaas no todas son de calidad, puesto que como se ha dicho, hay fallos en el proceso de secuenciación. Hay que delimitar dos extremos, tanto el margen izquierdo como el derecho. Si nos fijamos en la imagen primera de este trabajo, no se eligió al azar. Marca el paso de una zona de alto ruido (no es posible determinar bien los picos y otra zona muy limpia, donde claramente se puede definir las bases. Además, en el primer BLAST-n se nos confirma que en torno a la base 161-162 es la secuencia coincide, por lo que seleccionamos como límite izquierdo la base 162 en este caso. En cambio el otro margen no queda tan claro, ya que la región que marca el BLAST-n como límite es un tanto confusa en nuestro cromatograma como se puede ver a continuación.

Ahora hacemos otro BLAST-n, delimitando la secuencia tal y como llevamos hecho hasta ahora, buscando específicamente en Fragaria vesca. El resultado es el siguiente:

Sería complicado afirmar que esas bases son todas correctas en lo que a los picos respecta aún coincidiendo con el cromatograma, por lo que necesitamos un criterio para decidir. Este será la discrepancia que no podamos corregir porque haya duda en la decisión. Buscando entre estas, la que encontramos se localiza en la base 691, donde hay un hueco en la secuencia que tenemos, ya que la secuencia del GenBank hay cuatro guaninas, como se muestra a continuación.

En el cromatograma apreciamos lo siguiente:

Por tanto, no podemos afirmar cuál es correcta ya que no hay solo cuatro posibles guaninas, sino cinco, por lo que es aquí donde en un principio marcaremos el límite derecho, en la base 691, siendo esta la última que se queda justo antes de este problema.
Ahora es momento de corregir las N (bases que se han detectado pero no se les ha asignado una base), encontrando que no hay ninguna dentro del margen de la secuencia que hemos delimitado. Por ello acompañamos de una corrección como ejemplo. En este caso apreciamos que se corresponde a una guanina, pues vemos un pico de color negro que aunque es más bajo que los de su alrededor, se ve claramente. Por otra parte podemos comprobar en el BLAST-n

Podemos contrastar la información con el BLAST-n que hicimos, donde apreciamos que efectivamente, esa base es una guanina, de modo que habría mejorado la calidad del BLAST-n si tuviese en cuenta esta región. Se muestra la imagen del BLAST-n a continuación:

A lo largo de la secuencia encontraremos puntos donde haya dudas, ya que no todos los picos son tan claros como cabría esperar, mostrando el siguiente ejemplo:

Se aprecian 3 claros picos de guanina, pero hay uno que no queda completamente claro (marcado en azul) si es un pico y se corresponde con una base o es simplemente un hombro de la curva. Ante esta duda podemos adoptar dos soluciones: tomar la idea de que, según nuestro criterio hay una cuarta guanina o no, o la opción que vamos a tomar, irnos al BLAST-n y comparar si en esa posición hay o no 4 guaninas. En el mismo encontramos el siguiente resultado:

Vemos que la secuencia devuelta por la base de datos, solo hay tres guaninas y no cuatro como podríamos sospechar, por lo que esta es la base de la decisión a tomar: ese posible pico no es más que el hombro de la curva, por lo que no se corresponde con ninguna base. Es necesario indicar en este punto que la secuencia que usamos como referencia procede de otra secuenciación, por un tercero, por lo que no sabemos los errores que se pudieron cometer a la hora de hacer dicha secuencia, así pues aceptar lo que esta dice siempre entraña asumir cierto riesgo, que en este caso parece aceptable ya que nosotros no tenemos clara la respuesta a la pregunta.
Sin embargo, hay que analizar también la traducción de la secuencia, ya que el resultado final es el de una proteína, por lo que la similitud con esta puede aportar nueva información sobre la calidad del cromatograma. Al igual que en el caso anterior, podemos hacer un BLAST-x, el cual se hace sobre la proteína resultante. El resultado de muestra a continuación:

La secuencia está muy bien pero hay un punto marcado con un asterisco, que simboliza un codón de STOP, de modo que teóricamente si la secuencia fuese correcta veríamos que la proteína resultante no sería tan larga como la silvestre. Es necesario ir a cromatograma y analizar qué ocurre, si podemos mejorar lo que tenemos o cambiar el corte y asegurar más. Lo que si que podemos afirmar que de los tres marcos de lectura posibles el correcto es el segundo, aunque, como esto depende de dónde hemos cortado podría haber discrepancia, por lo que comprobaremos cual es el correcto en el programa. Para ello hacemos que se muestren los tres marcos de lectura posibles como se ve en la imagen siguiente, correspondiente a la zona discrepante.
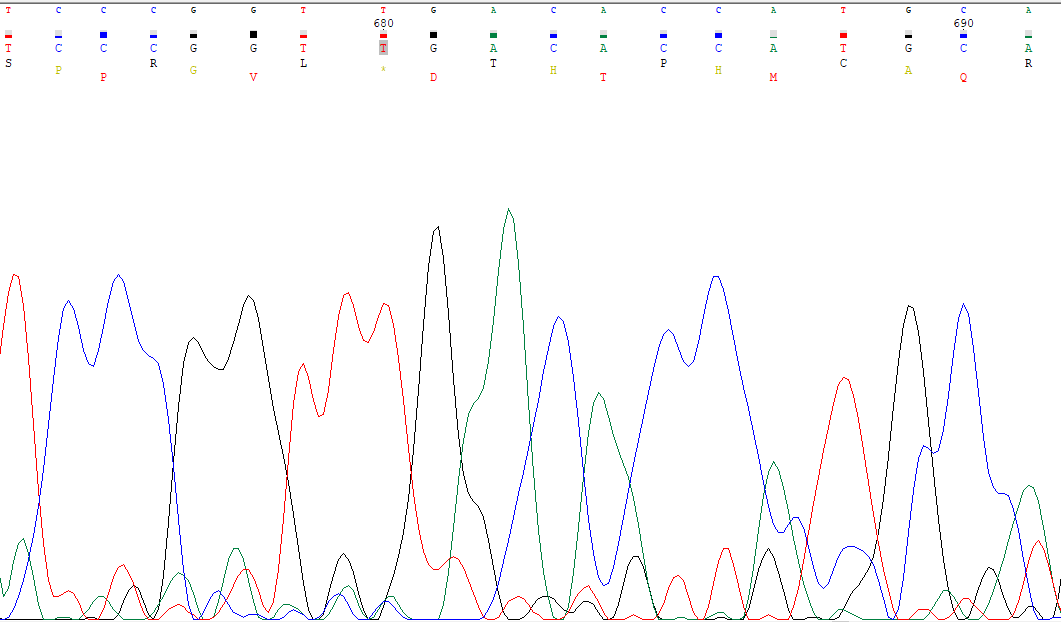
Como vemos, efectivamente el marco de lectura es el segundo, ya que se da la secuencia correcta. Por otro lado, se comprueba que es un codón de stop el que se codifica en esa zona (TGA). Una posible solución sería intentar evitar dicho codón de stop, para lo que analizamos qué podríamos cambiar (siempre congruente). Este posible cambio sería añadir el pico que, según parece, no se detectó. Si se hace dicho cambio, vemos que hay un desfase y por tanto la proteína resultante se ve alterada:

En consecuencia, esta zona muestra una indecisión clara, por lo que, es conveniente dejarla fuera de la región que consideramos es correcta. Así pues el cromatograma qudaría con el límite derecho en la base 674. Para comprobar si esto ha hecho que la calidad del BLAST aumente, repetimos ambos, tanto n como x, cuyos resultados mostramos a continuación.

En lo que al BLAST-n respecta, vemos como la calidad ha mejorado, pasando el porcentaje de identidad de 97 a 98% y los huecos (gaps) al 0%, de modo que la corrección es desde el punto de vista del BLAST-n muy buena.

Por otra parte, en el BLAST-x ocurre parecido, pasando del 94% de identidad al 98%, un claro salto de calidad. También vemos que las diferencias que hemos encontrado entre BLAST-n y secuencia del cromatograma que deducimos, no podíamos cambiar por lo definido del pico, son SNP )Single Nucleotide Polimorfism) si no superan el 1% de distribución entre la población o mutaciones, pero en todo caso, silenciosas ya que el cambio en el genoma no se ve reflejado en el fenotipo gracias a la degeneración del código genético, que amortigua las variaciones inducidas en el genoma.
No hay comentarios:
Publicar un comentario